
—任何真理都有其边界,任何认知都会随实践而深化
在药物研发领域,Lipinski于1997年提出的"类药五原则"(Rule of Five, Ro5)长期被视为指导小分子口服药物设计的经典准则。在过去数十年的药物开发实践中,这一规则体系为筛选具有成药性潜力的候选分子建立了重要的理论框架,诸多成功上市的药物均符合该原则所界定的参数范围。然而,随着现代药物化学将目光看向更广阔的方向——例如致病蛋白的彻底降解、缺乏深口袋的“不可成药”靶点——这套曾经指引方向的原则,是否仍然需要遵守?
成药性(Druggability)的底层逻辑是什么?
药物成药性是活性分子能否跨越临床成为真正药物的决定性属性。对于传统小分子而言,成药性的底层逻辑建立在静态的物理化学限制之上。Ro5要求分子的分子量(MW)≤ 500 Da、脂水分配系数(LogP)≤ 5、氢键供体(HBD)≤ 5、氢键受体(HBA)≤ 10。这些严格的数值本质上是对“溶解度”与“膜渗透性”的精妙妥协:太亲水则无法穿透细胞膜的脂质双分子层,太亲脂则会在水性环境中沉淀且极易被肝脏代谢。
在药物发现的早期,获得一个具有极高亲和力(Affinity)的化合物是关键的第一步,但高亲和力并不等于高药效,更不等于成药。药物在体内需要经历吸收、分布、代谢、排泄(ADME)的重重考验。如果忽视成药性边界,即使分子在体外能以皮摩尔级别死死咬住靶点,它也可能因为体积过于庞大、极性表面积(TPSA)超标而根本无法进入细胞,或者在进入血液的瞬间就被血浆蛋白紧紧结合而失去游离活性。优化成药性,本质上是在为亲和力搭建通往靶点的桥梁。
符合Ro5的经典药物案例:布洛芬和伊马替尼
布洛芬:布洛芬是非甾体抗炎药中应用最广的药物之一,其各项理化参数均显著低于Ro5的上限(Table 1)。分子量为206.3 Da,含1个氢键供体与2个氢键受体,拓扑极性表面积较小;LogP约为3.97,脂溶性良好。这些结构特征使其在体内可通过被动扩散快速穿透胃肠道黏膜及细胞脂质双分子层,口服吸收率接近100%,起效迅速。
布洛芬属于丙酸衍生物,结构可区分为亲水头部与亲脂尾部两部分(Figure 1)。在结构功能方面,羧基作为亲水头部是药物与靶点相互作用的关键位点,提供1个氢键供体与2个氢键受体,在体内进入环氧合酶(COX-1/COX-2)的活性口袋后,与Arg120和Tyr355残基形成离子键与氢键,从而抑制前列腺素的合成,发挥镇痛抗炎作用。异丁基苯基作为亲脂尾部为疏水性结构,可嵌入COX酶活性口袋的疏水通道中,通过范德华力增强结合稳定性。
在成药性方面,布洛芬的亲脂尾部与较小的分子量使其LogP值较高,被动渗透性较强。游离态布洛芬水溶性较低,但羧基结构使其可制备为钠盐或精氨酸盐等成盐形式,提高在胃肠液中的溶出速率,以游离酸形式快速吸收,从而实现较快的起效时间。
伊马替尼:伊马替尼是靶向治疗领域具有代表性的药物之一,其分子设计在符合Ro5框架的前提下体现了较高的结构复杂度。分子量为493.6 Da,接近500 Da的上限;氢键受体数量为7个。在结构优化的过程中,其LogP被控制在3.8左右,在水溶性与跨膜渗透性之间实现了平衡,使其在具备较高靶点亲和力的同时,口服生物利用度接近98%。
伊马替尼的分子结构可划分为多个功能模块,各模块分别与BCR-ABL激酶活性口袋的不同区域相互作用。嘧啶环与吡啶环参与与铰链区甲硫氨酸(Met318)的氢键结合;酰胺键作为连接部分,与谷氨酸(Glu286)及天冬氨酸(Asp381,位于DFG基序)形成氢键,将激酶稳定于无活性的构象;苯环上的甲基通过空间位阻影响分子构象,有助于提高对BCR-ABL激酶的选择性,减少对其他激酶的抑制作用。
在成药性优化方面,早期分子因芳香环较多导致水溶性不足,影响口服吸收。引入N-甲基哌嗪环后,该结构在生理条件下可质子化,显著提升水溶性,并将LogP调节至适宜范围。此外,哌嗪环使伊马替尼可与甲磺酸形成盐,进一步改善溶解度与制剂稳定性,最终实现较高的口服生物利用度。
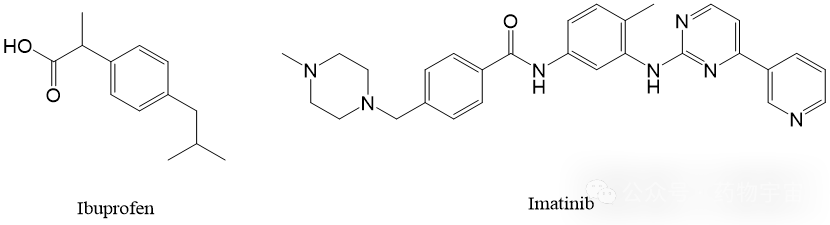
Figure 1 The structures of Ibuprofen and Imatinib.
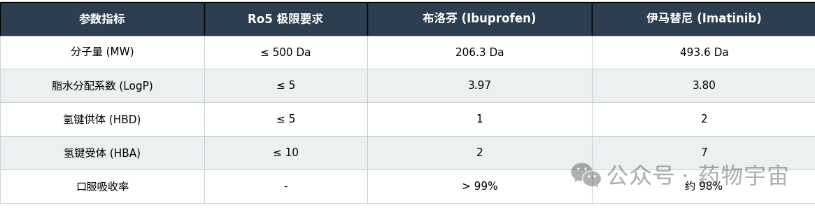
Table 1 Comparison of several indicators related to the Ro5 between Ibuprofen and Imatinib.
bRo5的出现:大环化合物和双功能分子设计
在药物研发领域,Lipinski的“类药五原则”(Ro5)长期以来是评估口服生物利用度的金标准。然而,随着未满足医疗需求的增加以及对难成药靶点的不断探索,药物化学家们正大步迈入“超越五原则”( Beyond the Rule of 5,bRo5)的化学空间。从大型复杂小分子到双功能分子(如PROTACs),突破传统参数限制并不意味着放弃成药性规则,而是需要建立全新的多维评价体系。
· 大环化合物
大环化合物是bRo5药物设计的一个新兴且重要的焦点 。在bRo5空间中,分子量、脂水分配系数、氢键供体和受体数量往往远超传统限制。即便如此,约6%的口服药物存在于Lipinski空间之外,这证明了bRo5药物的巨大潜力。为了更好地指导设计,AbbVie的研究人员基于其庞大的临床前药代动力学数据库提出了一种名为AB-MPS(AbbVie Multiparametric Score)的简单多参数评分系统(Figure 2)。该系统公式为:AB-MPS = Abs(cLogD - 3) + NAR(芳香环数量)+ NRB(可旋转键数量)。
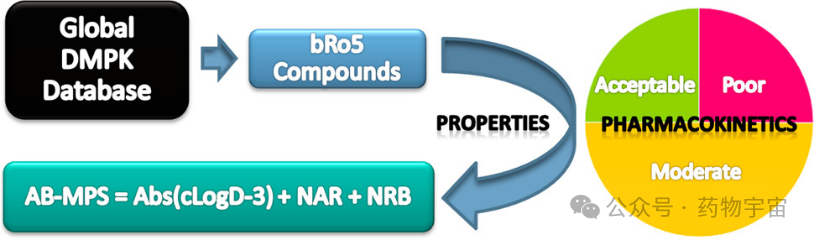
Figure 2 Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection.
DOI: 10.1021/acs.jmedchem.7b00717
随着分子量的增加,分子的柔性通常会急剧上升,导致巨大的构象熵罚,极大地阻碍跨膜吸收。AbbVie研发的这批HCV NS3/4A抑制剂体积庞大,分子量(MW)介于719.8至869.9之间,远超Ro5规定的500 Da上限。尽管分子量巨大,但得益于其大环骨架的刚性约束,这些化合物成功地将可旋转键数量(NRB)控制在了10个以内。这种“大而刚”的设计使得分子保持了“类药”的NRB水平,大大降低了穿过脂质双分子层时的构象能量损耗。
另一方面,极性表面积是评估渗透性的关键。对于传统小分子,拓扑极性表面积(TPSA)>140通常意味着极差的渗透性和口服吸收率。这批HCV大环抑制剂的计算TPSA值非常高,集中在175到225之间(Table 2)。如果仅看这个2D静态参数,它们几乎不可能口服成药。然而,研究人员通过超临界流体色谱测定的有效极性表面积(EPSA)揭示了真相。大环分子极易发生自组装行为,数据显示,这些分子的EPSA值大幅低于TPSA值。例如,化合物16的EPSA仅为89,完美落入了可接受渗透性的区间。这强有力地证明了这些大环分子能够在非极性环境(如细胞膜)中通过形成分子内氢键(IHB)将极性基团隐藏起来,展现出典型的“变色龙”特性(Chameleonicity)。
AbbVie团队提出的AB-MPS多参数评分公式在这个大环系列中得到了很好的验证。对化合物12-19进行回顾性计算,发现它们的AB-MPS值集中在12到14.5这样一个非常狭窄且有利的区间内。这一评分预测它们具有中等至高度的口服吸收潜力,这与体内药代动力学实验中观察到的高肠道吸收率高度一致。最终上市的同类大环药物(如grazoprevir和paritaprevir)的AB-MPS值也分别仅为10.5和12.2,进一步印证了该指标的可靠性。
Figure 2 Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection.
DOI: 10.1021/acs.jmedchem.7b00717
· 双功能分子
对于双功能分子而言,成药性的底层逻辑转向了动态的构象适应性。双分子通常需要同时招募两个靶蛋白,其分子量往往在700至1300 Da之间,严重违反Ro5。为了实现口服给药,药物化学家必须在看似不可调和的高水溶性与高膜渗透性之间寻找平衡。bRo5药物设计的底层逻辑,转向了构象灵活性与分子内非共价相互作用的动态调控,而连接子正是实现这一调控的关键。这篇发表于《Nature Reviews Chemistry》(2025年)的综述文章《Lessons learned in linking PROTACs from discovery to the clinic》由阿斯利康(AstraZeneca)的研发团队撰写 。文章系统性地总结了双功能降解剂(特别是PROTACs)从早期发现到临床转化过程中的经验教训,重点剖析了连接子在优化药理学、提升口服生物利用度以及改善代谢命运中的决定性作用 (Figure 3)。
Figure 3 Role of the linker in optimizing pharmacology, oral bioavailability, and metabolic fate.
DOI: 10.1038/s41570-025-00784-6.
“变色龙效应”是庞大的PROTAC分子跨越细胞膜的底层逻辑 。PROTAC 能够在不同的溶剂环境中动态调整其三维构象 :在极性的水相环境(如胃肠液或血浆)中,分子倾向于展开并暴露极性基团以增加溶解度;而在接触亲脂性的细胞膜时,分子则通过分子内氢键(IMHB)或疏水塌陷发生折叠,将氢键供体(HBD)“隐藏”起来,从而显著提升被动渗透性。
围绕这一效应,临床候选分子的优化遵循两项关键策略:
严格控制暴露的氢键供体:尽管 Ro5 建议小分子 HBD ≤ 5,但多项独立研究一致表明,PROTAC 的成药性优化需将溶剂可及的 HBD 数量严格控制在 2 个或以下。例如,将酰胺键替换为酯键,或通过空间设计形成分子内氢键来屏蔽 HBD,是提升口服吸收率的有效手段。
区分 HBD 的功能类型:临床阶段的口服 PROTAC(如 ARV-766)通过 NMR 分析发现,其结构中既包含能动态隐藏的“变色龙型 HBD”,也包含部分仅用于提升溶解度的“强溶剂化 HBD”。这种功能分化为高分子的双参数优化提供了更精细的设计维度。另一方面,与传统小分子抑制剂的静态结合不同,PROTAC 依赖于形成“目标蛋白(POI)-PROTAC-E3酶”三元复合物来驱动降解 。这种事件驱动的药理学在成药性评价上引入了新的维度:
正协同性(Positive Cooperativity,α> 1): 协同性是指PROTAC形成三元复合物的倾向性相对于形成无活性二元复合物的比例 。高度正协同性的PROTAC不仅能大幅提升降解效力,还能有效抑制或消除高浓度下容易出现的“钩状效应”(Hook effect,即高浓度下非生产性二元复合物占据主导,导致降解率下降)。围绕正协同性的优化,连接子的刚性化改造成为关键策略。早期靶点探索阶段,柔性连接子(如 PEG 链)常被用于提高形成三元复合物的概率,以快速验证降解活性。然而,在向临床候选药物优化时,通过引入芳香环等刚性结构来限制连接子的构象熵,往往能显著增强三元复合物的稳定性与协同性,将分子从“可降解”提升至“可口服”的成药水平。
回顾目前已披露结构的20款进入临床试验的PROTAC分子,绝大多数已进入临床的口服 PROTAC 均选择了 CRBN 作为招募的E3连接酶 。这并非巧合,而是因为 CRBN 配体(如来那度胺衍生物)分子量极小,且通常仅携带 1 个必须的氢键供体(HBD),这为庞大的双分子节省了极其宝贵的物理化学参数空间,使其更容易达到口服吸收的门槛 。相比之下,基于VHL的PROTAC由于配体较大且含有至少两个HBD,目前进入临床的分子均为静脉注射给药 。我们不妨将目光转向ARV-471(Vepdegestrant),一款靶向雌激素受体(ER)的PROTAC降解剂,主要用于治疗ER阳性乳腺癌,目前已进入临床III期试验(Figure 4)。首先,ARV-471选用了基于来那度胺的CRBN连接酶配体,为庞大的双功能分子节省了宝贵的理化参数空间。在bRo5空间中,严格限制暴露的HBD数量(通常建议≤2个)是提升跨膜渗透性和口服吸收的核心法则。其次,ARV-471采用的“哌嗪-哌啶”刚性组合,不仅消除了易被代谢酶识别的柔性脆弱位点,还通过限制构象,帮助分子在体内形成有利于跨膜吸收的特定形状。X射线晶体学和溶液NMR结构表明,ARV-471的刚性Linker显著限制了分子的构象熵,使其在维持降解活性的同时,获得了足以支持口服给药的物理化学性质。
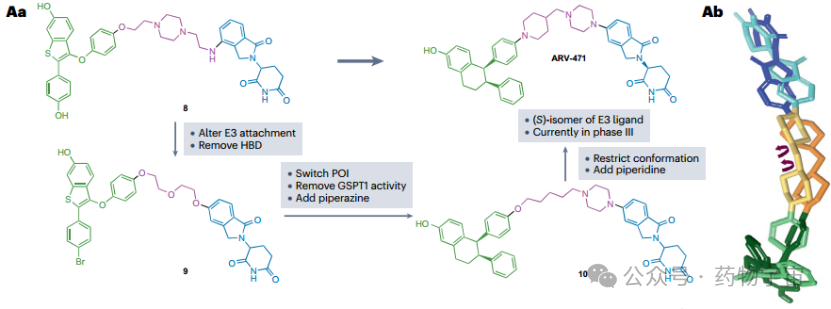
Figure 4 Development of ARV-471 and Three-dimensional superposition of the crystal NMR structure (dark colours) and solution NMR structure (light colours).
DOI: 10.1038/s41570-025-00784-6.
思考与展望
回顾这段从Ro5到bRo5的发展史,我们可以清晰地看到,药物研发从来不是为了迎合某项纸面上的生硬规则,而是为了解决真实的临床痛点。未来的药物设计本质上是一个复杂的多因素优化过程。无论是借助AB-MPS评分、EPSA等工具预测吸收概率,还是利用冷冻电镜和分子动力学模拟调控三元复合物构象,其核心始终是在极性与亲脂性之间寻求平衡,在柔性探索与刚性协同中实现药效优化。
这一范式转变反映了药物化学从遵循经验规则转向理解规则背后的物理化学原理,并在此基础上设计补偿机制。Ro5的价值在于指出了小分子口服吸收的关键限制因素——分子量、氢键供体、可旋转键——使得在突破这些限制时,必须明确相应的补偿策略。大环化合物和双功能分子设计是这些策略最好的先行者。
Ro5并非不可逾越的边界,bRo5则代表了对成药性边界的进一步拓展。这一边界的延伸,依赖于在具体情境下对规则的合理应用与突破,并以临床价值为最终判断依据。
参考资料:1. DeGoey DA, Chen HJ, Cox PB, Wendt MD. Beyond the Rule of 5: Lessons Learned from AbbVie's Drugs and Compound Collection. J Med Chem. 2018;61(7):2636-2651. 2. García Jiménez D, Rossi Sebastiano M, Vallaro M, et al. Designing Soluble PROTACs: Strategies and Preliminary Guidelines. J Med Chem. 2022;65(19):12639-12649.3. Pike A, Lee ECY, Michaelides IN, et al. Lessons learned in linking PROTACs from discovery to the clinic. Nat Rev Chem. 2026;10(2):117-132. 4. Han X, Wang C, Qin C, et al. Discovery of ARD-69 as a Highly Potent Proteolysis Targeting Chimera (PROTAC) Degrader of Androgen Receptor (AR) for the Treatment of Prostate Cancer. J Med Chem. 2019;62(2):941-964.往期回顾:亲和力:药物设计的关键?蛋白多聚:分子新设计?eTPD 胞外降解:跨血脑屏障的药物设计LYTAC:拓展降解剂的边界CPPTACs: 膜蛋白降解









